Lanthanide elements (Ln), as a crucial component of rare earth elements, represent significant strategic resources for China. In recent years, investigating the applications of lanthanides in the new energy sector has emerged as a vital strategic direction for rare earth materials. With the rapid development of the new energy industry, the performance requirements for lithium-ion batteries have continuously increased. One of the primary approaches to enhancing their performance lies in the optimization and modification of cathode materials. Currently, LiNiO2 (LNO) has attracted growing attention due to its advantages of high energy density and low cost. However, challenges such as chemical stability, structural stability, and thermal stability remain to be thoroughly studied and addressed.
A commonly effective approach for optimizing and modifying LNO cathode materials is elemental doping. Lanthanide elements possess distinctive characteristics that make them promising dopants for LNO, including: 1) relatively large ionic radii, 2) strong bonding affinity with oxygen, and 3) electrochemical inertness. However, the research on LNO+Ln faces dual challenges. Experimentally, the large ionic size of lanthanides makes their doping into layered structures particularly difficult. Computationally, systems containing lanthanide elements suffer from high computational costs, poor convergence behavior, and unstable calculation results.
Recently, Prof. Zheng Jiaxin’s group conducted extensive first-principles calculations on LNO+Lanthanide (Ln) systems to systematically investigate their electronic structure characteristics. Their study revealed that the diverse configurations and strong correlation effects of 4f electrons in high-spin states induce multiple metastable states in LNO+Ln systems, which significantly compromises computational convergence and result accuracy. Building upon this fundamental understanding, the team developed a ground-state search method that enables rapid, stable, and precise calculations for LNO+Ln systems. These significant findings were published in SCIENCE CHINA Materials (JCR Q1, Top) under the title “Ground-State Search and Modification Effects of Lanthanide Substitution in LiNiO2: A First-Principles Study”.

Figure 1. Schematic illustration of metastable states and ground state in lanthanide element theoretical calculations.
The accuracy and stability of ground-state electronic structure calculations in theoretical models are intrinsically linked to their magnetic configurations. Conventional computational models typically converge to either high-spin or low-spin states during calculations, which are readily distinguishable due to their significant differences in both energy and magnetic moment values. Prof. Zheng Jiaxin’s group discovered that in LNO+Ln systems, the high-spin state exhibits lower energy. Notably, unlike conventional systems, LNO+Ln systems demonstrate a peculiar phenomenon where minute differences in magnetic moments can induce substantial energy variations of approximately 1 eV. This behavior originates from the existence of numerous metastable states near the ground-state electronic structure of LNO+Ln systems. These metastable states exhibit magnetic moment values nearly identical to the ground state (with only marginal differences), yet correspond to distinct 4f-electron configurations that result in considerable energy disparities. Consequently, during first-principles calculations, the computational models exhibit a tendency to converge to these metastable states rather than the true ground state. This leads to poor computational convergence and inaccurate simulation results, presenting significant challenges for theoretical investigations of LNO+Ln systems.
To address this unique theoretical challenge specific to rare-earth elements, Prof. Zheng Jiaxin’s group proposed an innovative ground-state search methodology. This methodology involves adjusting the initial magnetic convergence parameters to conduct low-cost, exhaustive self-consistent field (SCF) calculations under the DFT+U framework. A crucial finding reveals a definitive correlation between the converged state energy and the 4f-electron magnetic moment values, enabling precise identification of the electronic ground state through magnetic moment characterization. The developed method demonstrates three significant advantages over conventional computational approaches:
1. Substantially reduced computational resource requirements;
2. Remarkably enhanced calculation stability and efficiency;
3. Guaranteed high-precision results.
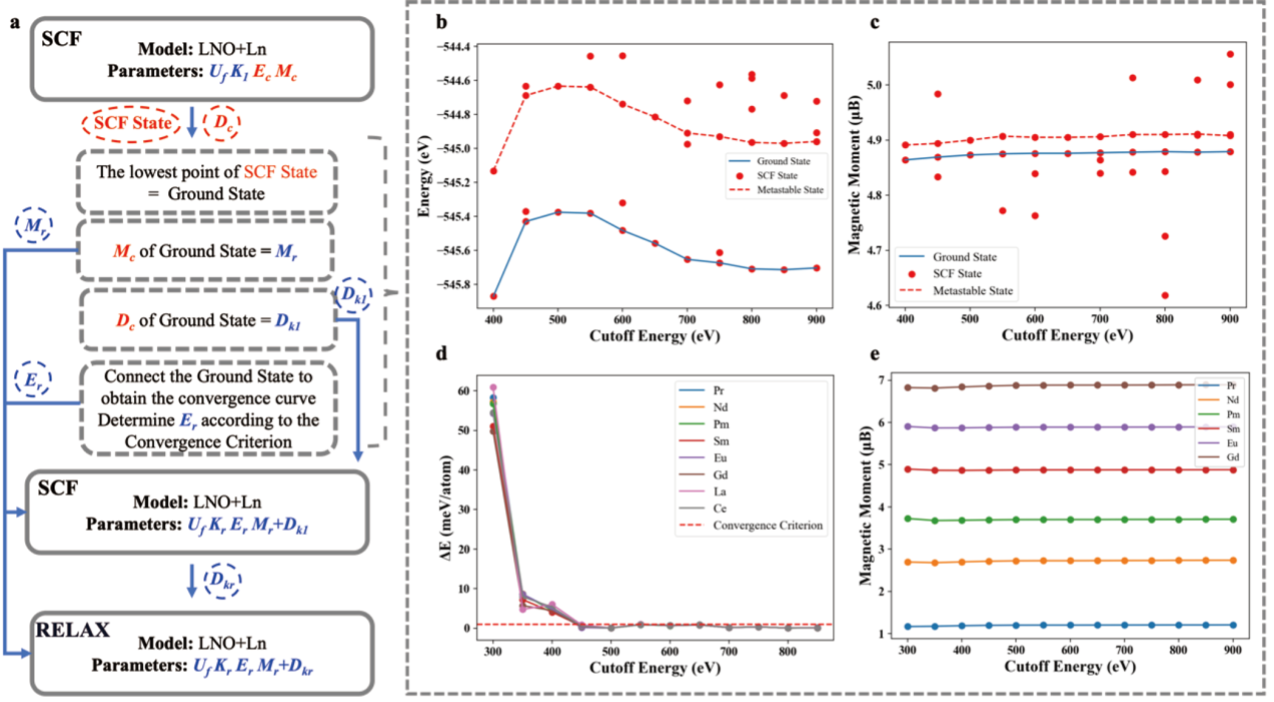
Figure 2. The process and test performance of the ground state search method for LNO+Ln (La-Gd)
Employing the ground-state search method, the research team efficiently obtained the precise ground-state structures (including both crystal and electronic structures) of LNO+Ln (La-Gd) systems and further calculated the Li/Ni disordering formation energies and oxygen vacancy formation energies. The study revealed that Ln doping in LNO significantly enhances its layered ordering, effectively suppresses Li/Ni disordering, and improves oxygen stability. These findings are in excellent agreement with recently published experimental studies on high-nickel LNO materials, validating the accuracy and reliability of the proposed methodology.
Regarding the mechanistic analysis, the team elucidated that Ln doping primarily suppresses Li/Ni disordering through ionic size effects, superexchange interactions, and oxidative environment effects. And the combined effects of Ln-O bond lengths, Ln-O bond strengths, and charge transfer collectively improve oxygen stability. The comprehensive study demonstrated that among all lanthanide elements, Ce doping exhibits the most outstanding performance, emerging as the most promising candidate for practical applications. The relevant computational results are presented in Figure 3.

Figure 3. Calculation results of doping modification of LNO+Ln (La-Gd)
Prof. Zheng Jiaxin and Dr. Ye Yaokun from School of Advanced Materials are the corresponding authors of this work. Wu Guangyin, a 2022-grade master student from School of Advanced Materials, is the first author. This work was supported by the National Natural Science Foundation of China and the Peking University Shenzhen Graduate School-Eacomp Technology Joint Laboratory of Battery Material Simulation.










