Electrocatalysis is a key technology for achieving sustainable energy conversion and carbon emission reduction. Currently, the dual challenges of high-concentration nitrate pollution in industrial wastewater and the enormous energy consumption of traditional Haber–Bosch ammonia synthesis pose serious environmental and energy problems. Electrocatalytic nitrate reduction reaction (NO3RR) offers a promising solution by converting nitrate into high-value ammonia under ambient conditions. However, NO₃RR involves a complex 8–9 electron-transfer process and suffers from severe competition with the hydrogen evolution reaction (HER), which severely limits Faradaic efficiency and practical applicability. Therefore, elucidating interfacial reaction mechanisms at the atomic level is crucial for the rational design of high-performance NO3RR electrocatalysts.
Professor Feng Pan’s group at the SAM, Peking University Shenzhen Graduate School, has long focused on solution structures and interfacial solvation environments. By systematically investigating interfacial composition, dynamic evolution, and local electric fields, the team has previously proposed several innovative mechanisms, including ordered interfacial water accelerating HER (Nature 2021, 600, 81–85), cation size effects at the interface governing C₂₊ selectivity in CO₂ reduction (J. Am. Chem. Soc. 2024, 146, 5532–5542), and interfacial *H spillover dramatically boosting nitrate reduction kinetics (J. Am. Chem. Soc. 2024, 146, 26965–26974).
Building on these foundations, Professor Feng Pan’s team collaborated with Professor Jianfeng Li’s group at Xiamen University and, using single-crystal gold electrodes as a model system, combined in situ spectroscopy with multi-scale theoretical simulations to reveal—for the first time—a synergistic enhancement mechanism between interfacial water networks and ion layering during electrocatalysis. The work, entitled “Elevating Nitrate Reduction through the Mastery of Hierarchical Hydrogen-Bond Networks”, has been published in the JACS (J. Am. Chem. Soc. 2025, 147 (24), 20504.)

Figure 1. Structure Transformation of Solution Concentration
Using in situ shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) and multi-precision molecular dynamics simulations, the team systematically investigated interfacial water structure and nitrate reduction kinetics on Au single-crystal electrodes in LiNO3 aqueous electrolytes across a wide concentration range (Fig. 1). They discovered that in concentrated electrolytes, NO3⁻ adsorbs parallel to the surface, forming an ordered Li⁺·NO3⁻ planar layer that facilitates activation of adsorbed NO3⁻. Simultaneously, vertically oriented NO3⁻ ions form extensive hydrogen bonds with surrounding water, constructing a highly connected interfacial hydrogen-bond network that dramatically accelerates NO3RR. Furthermore, by tailoring electrode facet orientation and electronic structure, the water–ion network can be further optimized to boost performance. These findings uncover the intrinsic correlation between water–ion interactions and hydrogen-bond network connectivity, demonstrating that strategic regulation of these factors offers a powerful new approach for optimizing complex interfacial reactions.
With SHINERS, the team achieved real-time observation of interfacial water structure on atomically flat Au(hkl) electrodes. Contrary to conventional wisdom that attributes enhanced H2On₌2 signals to Li⁺ enrichment, the study reveals that NO3⁻ anions play a dual role: acting both as hydrogen-bond acceptors and as structural bridges that dramatically improve network connectivity.
To gain atomic-level insight, the team constructed electrode/electrolyte interface models using classical molecular dynamics (MD) and ab initio molecular dynamics (AIMD). At optimal concentration and applied potential, a unique double-layer ordered structure emerges: The inner layer consists of an ordered 3 Li⁺·NO3⁻ plane, in which Li⁺ coordination weakens the N–O bond and lowers the activation energy; The outer layer contains vertically aligned NO3⁻ that serve as hydrogen-bond acceptors, substantially increasing the proportion of H2On₌2 species. This mechanism is highly concentration-dependent: it fails in dilute solutions due to insufficient ions and in overly concentrated solutions due to Li⁺ hydration chains locking the water network (Fig. 2).
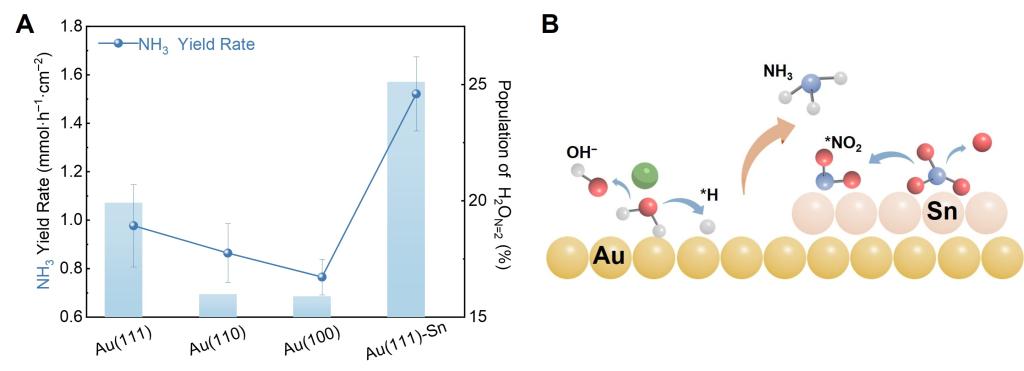
Figure 2. NO3RR performance of the Au(hkl) and Au(111)-Sn interfaces. (A) NH3 production rate and the corresponding H2O content at the 1:11 Au(111), Au(110), Au(100), and Au(111)-Sn/LiNO3 interfaces at −1.6 V. (B) Schematic diagram of the NO3RR process at the Au(111)-Sn interface.
Based on this mechanism, two electrode optimization strategies were proposed and validated: Facet engineering of single-crystal electrodes: different facets exhibit distinct potentials of zero charge (PZC). Au(111), with the most positive PZC, generates the strongest surface electric field at negative potentials, promoting ordered arrangement of Li⁺ and NO3⁻. Ammonia production rates follow the order Au(111) > Au(110) > Au(100), perfectly correlating with their PZC values. Heteroatom decoration: spontaneous adsorption of Sn on Au(111) forms Au(111)-Sn interfaces that further increase H2On₌2 content, doubling proton delivery efficiency. Theoretical simulation show that adsorbed Sn redistributes surface electrons and significantly lowers the energy barrier for NO3⁻ dissociation.
In summary, by combining in situ spectroscopy with multi-scale simulations, this work reveals that in concentrated electrolytes a hierarchical interface forms: an inner 3Li⁺·NO3⁻ plane activates nitrate via cation coordination, while outer vertically aligned NO3⁻ constructs a highly connected hydrogen-bond network that accelerates proton transfer. Subsequent facet engineering and Sn modification further optimize local electric field and electronic structure, achieving substantially enhanced ammonia yield. The study establishes a quantitative structure–activity relationship linking “electrolyte concentration → interfacial water network connectivity (H2On₌2) → proton transfer efficiency” and proposes a bifunctional design principle: “ion planar layer activates reactants/hydrogen-bond network accelerates mass transport”. This work provides an atomic-scale theoretical framework for electrolyte engineering and electrode microenvironment design, offering critical guidance for nitrate resource recovery and green ammonia synthesis.
In this work, Professor Feng Pan of Peking University and Professor Jianfeng Li of Xiamen University served as co-corresponding authors. Dr. Ruoyu Zhou and Assistant Professor Shisheng Zheng (formerly a PhD student in Professor Pan’s group at Peking University) of Xiamen University are the co-first authors. This research was supported by the National Natural Science Foundation of China, the Shenzhen Science and Technology Program, the Guangdong Provincial Department of Education, the Xiangjiang Laboratory Project, and the Major Scientific Infrastructure Program of the Materials Genome Initiative Platform.
Link to the paper: https://pubs.acs.org/doi/10.1021/jacs.5c02540










