新材料学院2020年6月23日讯,我院潘锋教授团队在研究工作中取得重要进展。
下一代电池要用更高能量密度的锂金属和钠金属作为锂电池和钠电池负极材料,要解决的关键科学和技术问题是如何控制和抑制金属的枝晶生长,因此从理论上研究金属晶体微观生长机理至关重要。金属晶体的生长模拟可分为两大类:一类基于第一性原理计算密度泛函理论(Density functional theory, DFT),另一类基于半经验原子间势场(Molecular dynamics force fields, FF),例如嵌入式原子方法(Embedded atom method, EAM)。这些方法的选择取决于计算精度和计算成本,一方面,使用第一性原理计算的优势是高精度,但由于其高昂的成本和O(N3)缩放比例,它通常限于数百个原子的大小;另一方面,使用半经验原子间势场计算数千个原子的系统相对容易,但是不能保证其准确性。半经验原子间势场适合宏观特性,例如固液转变温度,但是原子水平的微观力可能是不正确的,当与实际相比时,可能导致错误的微观生长机理。
故而介于第一性原理计算和半经验原子间势场计算之间,近日,北京大学深圳研究生院新材料学院潘锋团队和美国劳伦斯伯克利国家实验室汪林望团队联合提出了一种借助神经网络(Neural network, NN)来模拟钠金属晶体生长的方法(SANNP,全称Single atom neural network potential),该模型创新性地加入DFT能量分解的方法,计算体系中单个原子的能量,与其他机器学习方法相比,能量分解的方法可以通过相同的DFT计算数据获得更多信息,以达到计算结果既精确而又迅速的目的,训练步骤示意图。将这种方法用于金属钠原子系统,仅需要1000步DFT分子动力学数据足以训练准确的模型。在测试集中,DFT模型和SANNP模型关于原子能量和力的比较。该工作近日以”Neural Network Force Fields for Metal Growth Based on Energy Decompositions”为题,发表在物理化学材料领域知名期刊 (J. Phys. Chem. Lett. 2020, 11, 1364−1369, Nature Index )上。
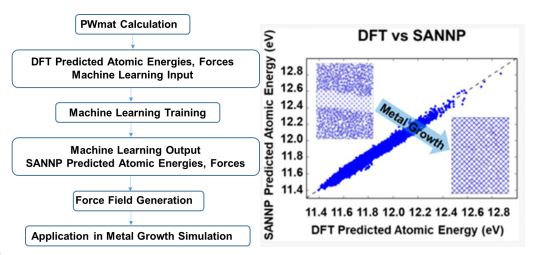
图1 神经网络力场模拟构建步骤示意图;DFT模型和SANNP模型比较
文章同时比较了DFT和SANNP的动力学参数,以验证其产生相似的结构特性。最后,在一个更大的系统中,模拟金属从液态到固态的生长过程,以证明SANNP模型具有模拟真实生长过程的能力,见图。作者期望类似SANNP模型的神经网络力场将来可以成为研究金属非平衡动力学行为的有力工具。
该工作由潘锋和汪林望指导完成,论文第一作者为硕士生胡钦,潘锋教授和汪林望教授为共同通讯作者。该项工作得到国家材料基因工程重点研发计划、广东省重点实验室项目、深圳市科技创新委员会项目、美国能源部材料理论项目等大力支持。
链接:https://pubs.acs.org/doi/10.1021/acs.jpclett.9b03780?ref=pdf




